Pharmaceuticals
Specialized language solutions for pharma
Specialized translation and language solutions to support your organization from molecule to global markets
Linguamatics' pre-set and proven clinical translation delivery model enables faster recruitment of participants, quicker data analysis, and more timely regulatory submissions, ultimately expediting the development and approval of life-saving treatments and therapies. Available on demand and in any language.
Packaging, labelling, product information and all other documents for regulatory submission handled through a centralized portal in any language and for any country. Our subject matter experts are always by your side to bring best practices and continuous optimization.

Roll down your campaign anywhere, anytime, and keep total control of your brand guidelines, while maintaining full compliance with local regulations. Raise engagement of HCPs by accounting for their language and culture, while simplifying your global operations and reducing costs.
Onboard all employees who require translations into a safe and data compliant environment, while reducing rising costs in managing multilingual content. Linguamatics offers solutions for all support function including Legal, HR, investor relations and corporate communications.

Linguamatics technology (suite: hub, translate and NLP) is the only language technology suite specifically created for life-science applications, servicing all language needs pharmaceutical companies' needs so they can perform on the global stage.

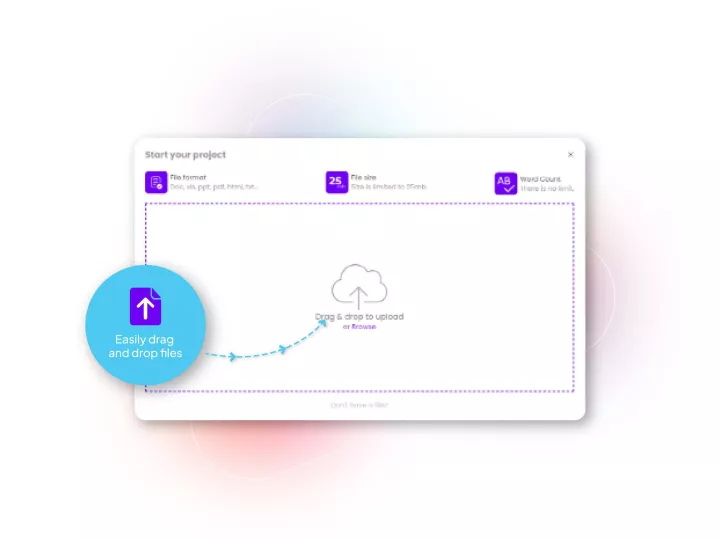
Linguamatics hub is our translation management system designed for Pharma’s unique needs in terms of compliance, speed and integration capabilities. It allows all your teams that require language solutions to obtain them in a few clicks, while maintaining strict compliance and reducing costs.
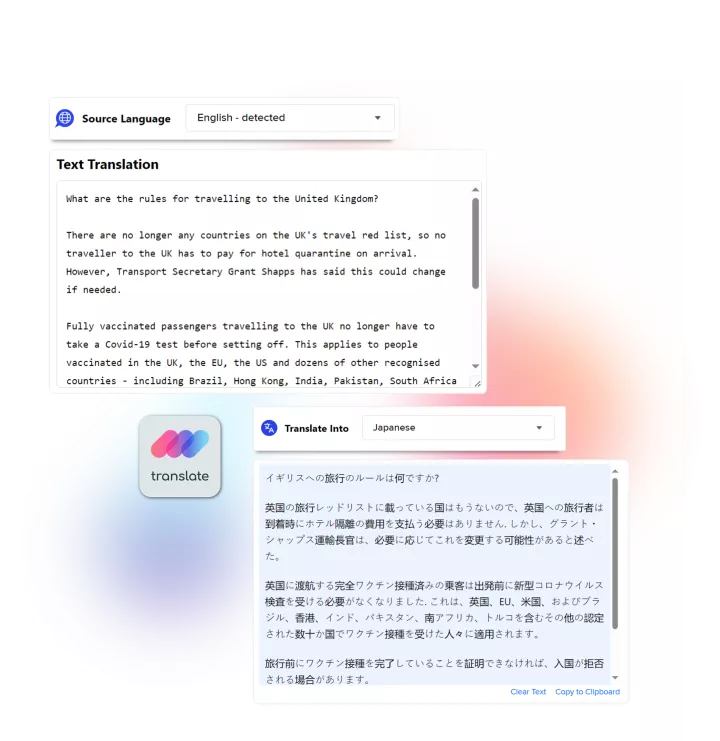
Linguamatics translate is a secured and compliant instant translation system allowing all your employee and systems to communicate in any language, securely:
Linguamatics NLP is our proprietary Natural Language Processing technology developed for life science use cases, and enabling unmatched efficiencies in drug development safety and pharmacovigilance, regulatory compliance and voice of the customer, in any language.